Nextflow is the the go-to tool for many people in the bioinformatics community who are working on developing data pipelines. Unfortunately, there is a pretty steep learning curve as there is a whole new Groovy-based syntax and framework for code organization to learn. The steepest part of this learning curve in my experience happened when I tried to move from a simple pipeline structure like those present in the main Nextflow documentation, to the fully-featured, best-practice templates used by the open-source nf-core community. To address this steep learning curve I've created a new, slimmed down Nextflow project template based off nf-core's main template. I hope it can be a stepping stone for intermediate Nextflow developers looking to learn best practices for pipeline development and for experienced Nextflow developers looking for a leaner codebase that can start generating outputs quicker than using the full template.
What is Nextflow?
Nextflow is a powerful workflow management tool that I frequently use to build, execute, and automate complex scientific pipelines. Its main strength lies in the ability to modularize virtually any program or custom code. Those modular of compute (called processes) can then be strung together into a workflow that can be run identically on a variety of computing infrastructures, including local computers, cloud-based platforms, and high-performance computing clusters. That modularity and portability are certainly two of the features that make Nextflow so popular among the bioinformatics community, but the most useful aspect of working with Nextflow is the open-source community that has developed around it.
nf-core is a community-driven project that provides a set of standardized Nextflow pipelines for some of the most common bioinformatics analyses. Some of these pipelines are very complex, so much so that they are visualized with metro map inspired diagrams like this one from nf-core/rnaseq:
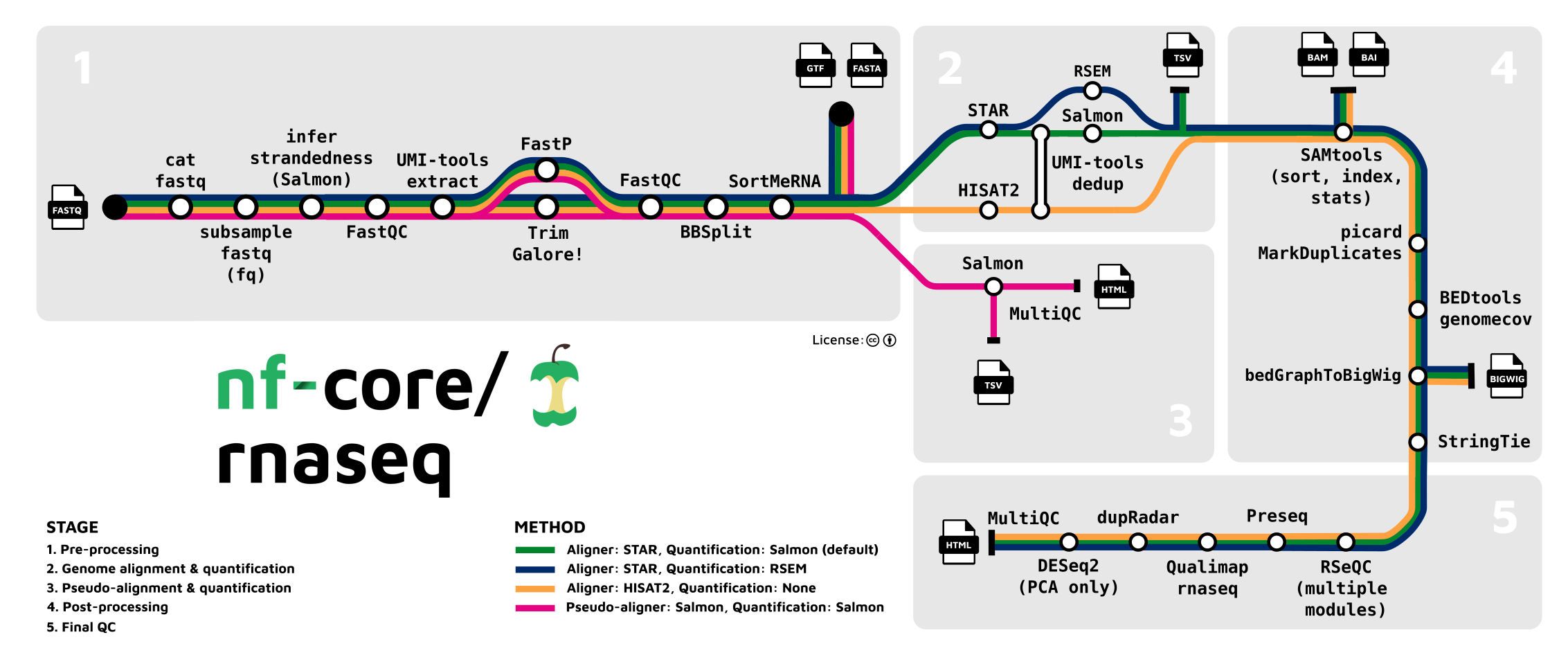
To manage these complex pipelines, nf-core has also developed a powerful python package called nf-core/tools that provides a set of command-line tools for creating, linting, testing, and syncing pipelines that adhere to nf-core standards.
Challenges of working with the default nf-core template
nf-core provides a template that can be used to create a new pipeline from scratch. This template is a great foundation for building a complex multi-step pipeline, but it can be overwhelming for beginning-to-intermediate Nextflow developers who are just trying to get a simple pipeline up and running while familiarizing themselves with Nextflow best practices. Even for experienced Nextflow developers, the nf-core template can be a bit of a pain to work with because of the many different areas of the template that need to be configured/modified to get some outputs for a new process.
A simpler nf-core-based template
To address some of the challenges of working with the default nf-core template, I created a simplenextflow template based on the nf-core template that is much simpler to work with. Here are some of the main changes I made:
Fewer files to search for relevant code
To ensure that the nf-core template is as flexible as possible, it is broken up into several different files that are used to configure various aspects of the pipeline. When I started developing pipelines using nf-core best practices, this was one of the most confusing aspects of the template. In simple-nextflow, I have moved the vast majority of configuration logic back into the nextflow.config file instead of having it imported from other files. Additionally, I have moved all of the workflow and subworkflow logic back into the main.nf file.
Instructions for adapting the pipeline
At the top of the README file, I also added some basic instructions of the core pieces of the pipeline that need to be modified in order to get change the template from the default fastqc example to a new pipeline.
Added config profile and templating for Wave containers
One of the most exciting new features of Seqera Labs has introduced is the ability to use Wave containers to run processes in a containerized environment without having to build a new container image for each process. Instead you can simply include a Dockerfile or environment.yml file in the process directory and Nextflow Tower will build a container image for that process on the fly. This is a great feature for developing pipelines because it allows you to quickly test out new code without having to build a new container image for each change. You can access this feature immediately in this template by running the pipeline with the -profile wave flag.
Removal of check_versions and MultiQC
The check_versions process and the MultiQC process are both great tools for ensuring that version information is captured accurately and you can visualize your results. However, they also fall in the category of features that are overwhelming for most new Nextflow developers. I've chosen to remove them from the template, to help make it easier to provide a simpler logic in the main.nf file. Best practices for capturing version information is still very important, and should be added back in for any pipeline that is intended for production use.
Keeping samplesheet logic
The concept of using a samplesheet to define the inputs was something that I considered cutting, because modifying the bin/check_samplesheet.py script to work with a new pipeline is one of the most time-consuming parts of adapting the template for a new pipeline. However, I think that setting up proper associations between files and their metadata is one of the most important practices in good pipeline development, so I decided to keep it in the template. I've been thinking about how to make the process of customizing the bin/check_samplesheet.py script easier, but I haven't come up with a good solution yet. Let me know if you have any ideas!
Things that are still in the template
Many of the wonderful quality of life features included in nf-core pipelines are still present in this slimmed down version. These are all features of the nf-core template that essentially work "out-of-the-box" with no additional configuration needed that would slow down the process to begin generating outputs. Some of my favorite features I was able to keep in the template are:
Reproducible development environments using Codespaces and Gitpod
Colorful logging and output of non-default parameters
Email and slack notifications
Conclusion
I hope this template is useful to anyone looking to familiarize themselves with nf-core best practices in more bite-sized chunks or are just looking for a simple template to get them to their desired outputs more quickly. Let me know if you have any suggestions for improving the template in the comments below or by making an issue/feature request in the GitHub repo.
References
Blog post changelog
- 2023-04-08 - Added new intro section



